
Published Work

O-Fucosylation plays crucial roles in various essential biological events. Alongside the well-established O-fucosylation of epidermal growth factor-like repeats by protein O-fucosyltransferase 1 (POFUT1) and thrombospondin type 1 repeats by POFUT2, we recently identified a type of O-fucosylation on the elastin microfibril interface (EMI) domain of Multimerin-1 (MMRN1). Here, using AlphaFold2 screens, co-immunoprecipitation, enzymatic assays combined with mass spectrometric analysis and CRISPR-Cas9 knockouts, we demonstrate that FUT10 and FUT11, originally annotated in UniProt as α1,3-fucosyltransferases, are actually POFUTs responsible for modifying EMI domains; thus, we renamed them as POFUT3 and POFUT4, respectively. Like POFUT1/2, POFUT3/4 function in the endoplasmic reticulum, require folded domain structures for modification and participate in a non-canonical endoplasmic reticulum quality control pathway for EMI domain-containing protein secretion. This finding expands the O-fucosylation repertoire and provides an entry point for further exploration in this emerging field of O-fucosylation.

Platelet activation induces the secretion of proteins that promote platelet aggregation and inflammation. However, detailed analysis of the released platelet proteome is hampered by platelets' tendency to preactivate during their isolation and a lack of sensitive protocols for low abundance releasate analysis. Here, we detail the most sensitive analysis to date of the platelet releasate proteome with the detection of >1300 proteins. Unbiased scanning for posttranslational modifications within releasate proteins highlighted O-glycosylation as being a major component. For the first time, we detected O-fucosylation on previously uncharacterized sites including multimerin-1 (MMRN1), a major alpha granule protein that supports platelet adhesion to collagen and is a carrier for platelet factor V. The N-terminal elastin microfibril interface (EMI) domain of MMRN1, a key site for protein-protein interaction, was O-fucosylated at a conserved threonine within a new domain context. Our data suggest that either protein O-fucosyltransferase 1, or a novel protein O-fucosyltransferase, may be responsible for this modification. Mutating this O-fucose site on the EMI domain led to a >50% reduction of MMRN1 secretion, supporting a key role of EMI O-fucosylation in MMRN1 secretion. By comparing releasates from resting and thrombin-treated platelets, 202 proteins were found to be significantly released after high-dose thrombin stimulation. Complementary quantification of the platelet lysates identified >3800 proteins, which confirmed the platelet origin of releasate proteins by anticorrelation analysis. Low-dose thrombin treatment yielded a smaller subset of significantly regulated proteins with fewer secretory pathway enzymes. The extensive platelet proteome resource provided here (larancelab.com/platelet-proteome) allows identification of novel regulatory mechanisms for drug targeting to address platelet dysfunction and thrombosis.

Intermittent fasting (IF) is an established intervention to treat the growing obesity epidemic. However, the interaction between dietary interventions and sex remains a significant knowledge gap. In this study, we use unbiased proteome analysis to identify diet-sex interactions. We report sexual dimorphism in response to intermittent fasting within lipid and cholesterol metabolism and, unexpectedly, in type I interferon signaling, which was strongly induced in females. We verify that secretion of type I interferon is required for the IF response in females. Gonadectomy differentially alters the every-other-day fasting (EODF) response and demonstrates that sex hormone signaling can either suppress or enhance the interferon response to IF. IF fails to potentiate a stronger innate immune response when IF-treated animals were challenged with a viral mimetic. Lastly, the IF response changes with genotype and environment. These data reveal an interesting interaction between diet, sex, and the innate immune system.
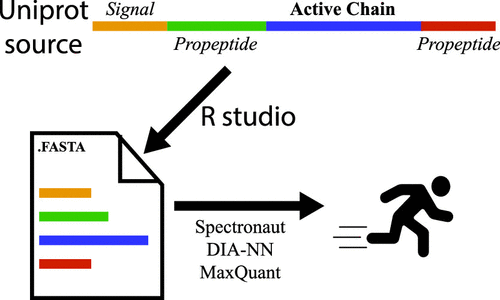
Liquid chromatography tandem mass spectrometry (LC–MS/MS) analysis of secreted proteins has contributed to our understanding of human disease and physiology but is limited by its need for accurate protein database annotation. Common assumptions used in proteomics of perfect protease specificity are inaccurate for secreted proteins, which are cleaved by numerous endogenous proteases. Here, we describe the generation of an optimized protein database that divides proteins into their individual biological chains and peptides to allow fast identification of semi-tryptic peptides from secreted proteins using fully tryptic searches. We applied this biologically annotated database to previously published human plasma proteome data sets containing either DIA or DDA data, using Spectronaut, DIA-NN, MaxDIA, and MaxQuant. Using our annotated database, we greatly reduced search times while achieving similar protein and peptide identifications compared to that obtained from standard approaches using semi-tryptic searches. Furthermore, our database enables the identification of biologically relevant semi-tryptic peptides using data analysis packages that are not capable of semi-tryptic searches. Together, these findings demonstrate that our annotated database is more capable than currently available databases for secreted protein analysis and is particularly useful for large-scale plasma proteome analysis.

Intermittent fasting is a beneficial dietary treatment for obesity. But the response of each distinct adipose depot is currently poorly defined. Here we explore the response of key adipose depots to every-other-day fasting (EODF) in mice using proteomics. A key change in subcutaneous white adipose tissue (scWAT) and visceral WAT (vWAT) depots is an increase in mitochondrial protein content after EODF. This effect is correlated with increased fatty acid synthesis enzymes in both WAT depots but not in brown adipose tissue. Strikingly, EODF treatment downregulates lipolysis specifically in vWAT, mediated by a large decrease in the abundance of the catecholamine receptor (ADRB3). Together, these changes are important for preservation of the visceral lipid store during EODF. Enrichment analysis highlights downregulation of inflammatory collagen IV specifically in vWAT, allowing improved insulin sensitivity. This resource for adipose-depot-specific fasting adaptations in mice is available using a web-based interactive visualization.

The increasing consumption of high-fat foods combined with a lack of exercise is a major contributor to the burden of obesity in humans. Aerobic exercise such as running is known to provide metabolic benefits, but how the over-consumption of a high fat diet (HFD) and exercise interact is not well characterized at the molecular level. Here, we examined the plasma proteome in mice for the effects of aerobic exercise as both a treatment and as a preventative regime for animals on either HFD or a healthy control diet. This analysis detected large changes in the plasma proteome induced by the HFD, such as increased abundance of SERPINA7, ALDOB, and down-regulation of SERPINA1E, CFD (adipsin). Some of these changes were significantly reverted using exercise as a preventative measure, but not as a treatment regime. To determine if either the intensity, or duration, of exercise influenced the outcome, we compared high-intensity interval training (HIIT) and endurance running. Endurance running slightly out-performed HIIT exercise, but overall, both provided similar reversion in abundance of plasma proteins modulated by the high-fat diet including SERPINA7, APOE, SERPINA1E, and CFD.

Every-other-day fasting (EODF) is an effective intervention for the treatment of metabolic disease, including improvements in liver health. But how the liver proteome is reprogrammed by EODF is currently unknown. Here, we use EODF in mice and multi-omics analysis to identify regulated pathways. Many changes in the liver proteome are distinct after EODF and absent after a single fasting bout. Key among these is the simultaneous induction by EODF of de novo lipogenesis and fatty acid oxidation enzymes. Together with activation of oxidative stress defenses, this contributes to the improvements in glucose tolerance and lifespan after EODF. Enrichment analysis shows unexpected downregulation of HNF4α targets by EODF, and we confirm HNF4α inhibition. Suppressed HNF4α targets include bile synthetic enzymes and secreted proteins, such as α1-antitrypsin or inflammatory factors, which reflect EODF phenotypes.
Small-protein enrichment assay enables the rapid, unbiased analysis of over 100 low abundance factors from human plasma.

Unbiased and sensitive quantification of low abundance small proteins in human plasma (e.g. hormones, immune factors, metabolic regulators) remains an unmet need. These small protein factors are typically analysed individually and using antibodies that can lack specificity. Mass spectrometry (MS)-based proteomics has the potential to address these problems, however the analysis of plasma by MS is plagued by the extremely large dynamic range of this body fluid, with protein abundances spanning at least 13 orders of magnitude. Here we describe an enrichment assay (SPEA), that greatly simplifies the plasma dynamic range problem by enriching small-proteins of 2-10 kDa, enabling the rapid, specific and sensitive quantification of >100 small-protein factors in a single untargeted LC-MS/MS acquisition. Applying this method to perform deep-proteome profiling of human plasma we identify C5ORF46 as a previously uncharacterized human plasma protein. We further demonstrate the reproducibility of our workflow for low abundance protein analysis using a stable-isotope labelled protein standard of insulin spiked into human plasma. SPEA provides the ability to study numerous important hormones in a single rapid assay, which we applied to study the intermittent fasting response and observed several unexpected changes including decreased plasma abundance of the iron homeostasis regulator hepcidin.
Proteomic Analysis of Human Plasma During
Intermittent Fasting

Intermittent fasting (IF) increases lifespan and decreases metabolic disease phenotypes and cancer risk in model organisms, but the health benefits of IF in humans are less clear. Human plasma derived from clinical trials is one of the most difficult sample sets to analyze using mass spectrometry-based proteomics due to the extensive sample preparation required and the need to process many samples to achieve statistical significance. Here, we describe an optimized and accessible device (Spin96) to accommodate up to 96 StageTips, a widely used sample preparation medium enabling efficient and consistent processing of samples prior to LC-MS/MS. We have applied this device to the analysis of human plasma from a clinical trial of IF. In this longitudinal study employing 8-weeks IF, we identified significant abundance differences induced by the IF intervention, including increased apolipoprotein A4 (APOA4) and decreased apolipoprotein C2 (APOC2) and C3 (APOC3). These changes correlated with a significant decrease in plasma triglycerides after the IF intervention. Given that these proteins have a role in regulating apolipoprotein particle metabolism, we propose that IF had a positive effect on lipid metabolism through modulation of HDL particle size and function. In addition, we applied a novel human protein variant database to detect common protein variants across the participants. We show that consistent detection of clinically-relevant peptides derived from both alleles of many proteins is possible, including some that are associated with human metabolic phenotypes. Together, these findings illustrate the power of accessible workflows for proteomics analysis of clinical samples to yield significant biological insight.
Efficient analysis of mammalian polysomes in cells and tissues using Ribo Mega-SEC

We describe Ribo Mega-SEC, a powerful approach for the separation and biochemical analysis of mammalian polysomes and ribosomal subunits using Size Exclusion Chromatography and uHPLC. Using extracts from either cells, or tissues, polysomes can be separated within 15 min from sample injection to fraction collection. Ribo Mega-SEC shows translating ribosomes exist predominantly in polysome complexes in human cell lines and mouse liver tissue. Changes in polysomes are easily quantified between treatments, such as the cellular response to amino acid starvation. Ribo Mega-SEC is shown to provide an efficient, convenient and highly reproducible method for studying functional translation complexes. We show that Ribo Mega-SEC is readily combined with high-throughput MS-based proteomics to characterize proteins associated with polysomes and ribosomal subunits. It also facilitates isolation of complexes for electron microscopy and structural studies.

REVIEW - Multidimensional proteomics for cell biology
The proteome is a dynamic system in which each protein has interconnected properties - dimensions - that together contribute to the phenotype of a cell. Measuring these properties has proved challenging owing to their diversity and dynamic nature. Advances in mass spectrometry-based proteomics now enable the measurement of multiple properties for thousands of proteins, including their abundance, isoform expression, turnover rate, subcellular localization, post-translational modifications and interactions. Complementing these experimental developments are new data analysis, integration and visualization tools as well as data-sharing resources. Together, these advances in the multidimensional analysis of the proteome are transforming our understanding of various cellular and physiological processes.